
por NeCiP | May 29, 2025 | Tumores de la región hipofisaria
Son un grupo heterogéneo de neoplasias caracterizadas por una gran variedad de tipos histológicos, pudiendo originarse en distintas zonas de la región Selar/hipofisaria. Los más frecuentes son: Craneofarigiomas, Tumores hipofisarios, Germinomas, y Hamartomas.
Los síntomas característicos son: cefalea, alteración de la agudeza visual, acompañado ocasionalmente de un síndrome de hipertensión intracraneana secundario a hidrocefalia.
Pueden provocar trastornos endocrinológicos ya sea por déficit o hipersecreción hormonal, sumado al efecto de masa. La alteración del eje hipotálamo-hipofisario y su repercusión sobre la secreción hormonal condicionan la aparición de obesidad, pubertad precoz, galactorrea y diabetes insípida. En raras ocasiones producen convulsiones y hemiparesia.
Si usted presenta estas manifestaciones clínicas, contáctese con nuestro equipo a la brevedad.
El diagnóstico se realiza con estudios complementarios, como son la tomografía (TC) y la resonancia magnética (RM). Siendo fundamental contar con equipos que permitan realizar estudios específicos y de alta complejidad (tomografía volumétrica, angioTC, angioRM, espectroscopía, RM funcional, tractografía, y neuronavegación, entre otros).
El tratamiento incluye varias opciones: la cirugía, la quimioterapia, la radioterapia y otras terapias.
La cirugía es la principal modalidad, para lograr el diagnóstico histológico (para determinar el tipo tumoral y definir el mejor tratamiento) y reducir el volumen tumoral.
Trabajamos en Centros de Salud con los mas modernos recursos tecnológicos para diagnóstico y tratamiento.
En la actualidad se están desarrollando terapias dirigidasque se proyectan como una nueva esperanza para el tratamiento de determinados tumores. Las denominadas vacunas contra el cáncer y la terapia génica son algunas de estas terapias, que buscan interferir en el crecimiento de células cancerosas específicas.
Nuestro equipo es pionero en técnicas mínimamente invasivas para el diagnóstico y tratamiento de los tumores cerebrales. Estando a la vanguardia y siempre en búsqueda de nuevas alternativas que permitan un mejor tratamiento y una cura definitiva.
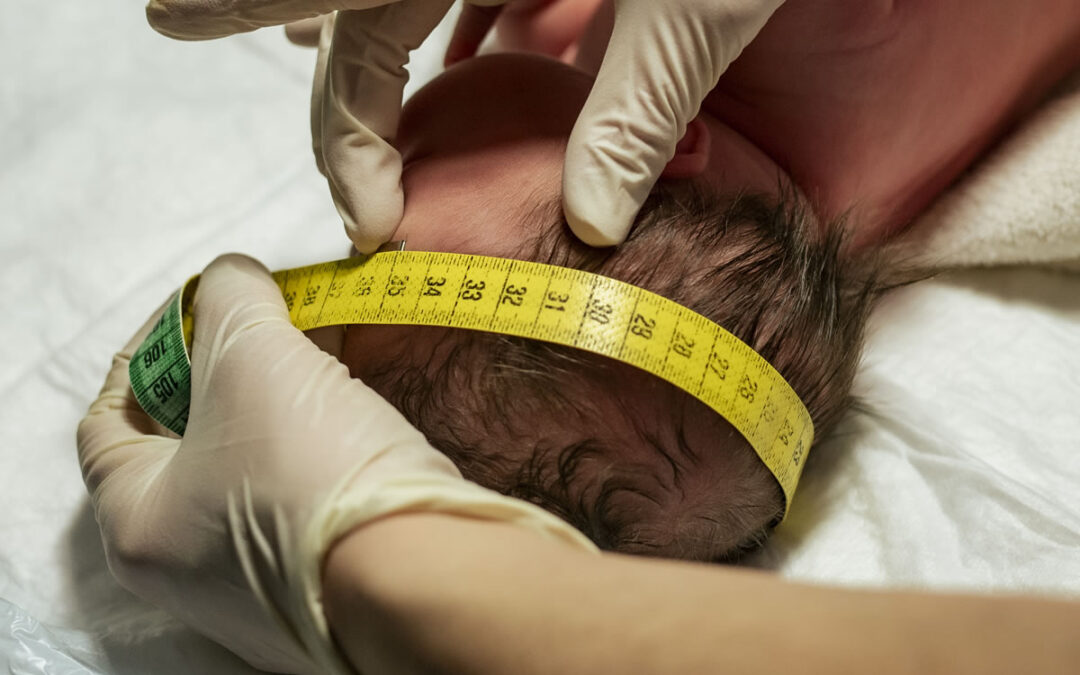
por NeCiP | May 29, 2025 | Craneosinostosis
Los distintos tipos de craneosinostosis son:
- Escafocefalia: cabeza alargada antero-posterior, frente prominente
- Plagiocefalia anterior: aplanamiento de la frente y órbita de un lado
- Braquicefalia: aplanamiento de la frente y órbita de ambos lado
- Trigonocefalia: frente con quilla triangular
- Plagiocefalia posterior verdadera: aplanamiento del craneo posterior
- Turricefalia/oxicefalia
- Cráneo en trébol
En cuanto a la clínica los pacientes con craneosinostosis simple en general son asintomáticos, sin clínica de hipertensión endocraneana, ya que el volumen total del cráneo es normal. De todas formas, pueden presentar algún déficit cognitivo. Los pacientes con craneosinostosis complejas pueden presentar déficit cognitivos, trastornos visuales y clínica de hipertensión endocraneana.
El diagnóstico principalmente es clínico, pero se completa con radiografías de cráneo como primer estudio, si bien en casos dudosos o complejos es necesario realizar la TC de alta resolución con reconstrucción 3D. Esta permite descartar malformaciones asociadas y realizar un análisis global de las estructuras craneo-faciales.
El tratamiento, generalmente, es quirúrgico variando entre distintas técnicas para obtener la mejor remodelación y reconstrucción craneo-facial, de forma individual en cada paciente.
Esto nos permite obtener un óptimo resultado neuroquirúrgico, funcional y estético.
La Plagiocefalia posterior posicional (no verdadera), no presenta un cierre precoz de la sutura. El tratamiento es no quirúrgico, y consiste en el cambio de decúbito del bebe o eventualmente en una ortesis externa (casco craneal remodelador). Se logra un buen resultado cuando el tratamiento se instaura dentro del primer año de vida del bebe.
Ante cualquier duda, contáctese con nuestro equipo a la brevedad.
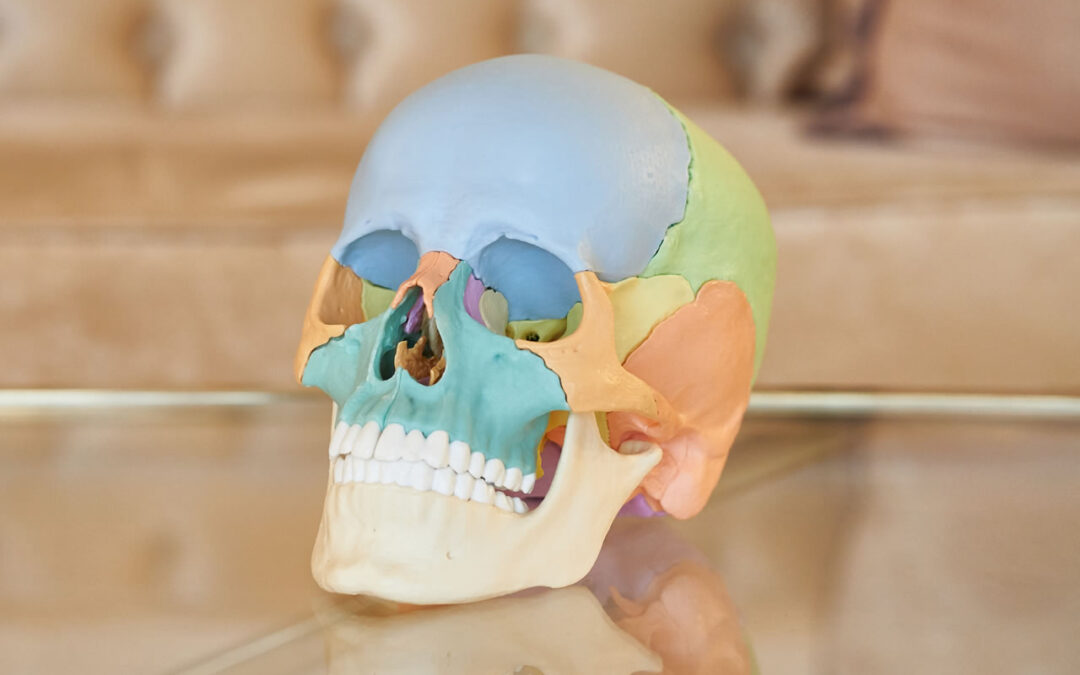
por NeCiP | May 28, 2025 | Sindromes craneofaciales
Los pacientes presentan rasgos faciales y craneanos característicos según el síndrome, lo que lleva a un diagnóstico temprano. Es necesario un enfoque multidisciplinario para su tratamiento.
Incluyen un grupo de patologías que pueden comprometer la bóveda craneana, las órbitas, el macizo facial y la base de craneo, con variada complejidad.
En nuestro equipo los pacientes son evaluados junto con cirujanos plásticos craneo–faciales especializados, con el objetivo de obtener el mejor tratamiento neuroquirúrgico, funcional y estético.
por NeCiP | May 28, 2025 | Defectos del cierre del tubo neural
(Espina bífida: Mielomeningocele; Siringomielia, Médula anclada)
Pueden ser espinales, afectando mayormente la región lumbosacra, o craneanos dependiendo de la zona afectada.
Los disrafismos (falta de cierre) espinales se dividen en dos grupos: espina bífida abierta (defecto en la piel, sumado al óseo, muscular, dural y en tejido nervioso) y espina bífida oculta (el defecto se encuentra cubierto por piel sana).
En general, son diagnosticados en ecografías fetales, pudiendo ser necesario una resonancia magnética fetal. Al nacimiento, se completan estudios diagnósticos para determinar el compromiso y mejor tratamiento.
Los disrafismos espinales son una causa de discapacidad y tienen que ser manejados por un equipo multidisciplinario especializado (clínico/pediatra, urólogo, traumatólogo, psicólogos, neurocirujano especializado, entre otros).
Ante cualquier duda, contáctese con nuestro equipo.
Dentro de las espinas bífidas abiertas el mielomeningocele (MMC) es la forma más común de presentación. Se caracteriza por la falta de fusión del tubo neural en su porción distal, con la consiguiente extrusión y exposición de la placa neural y nervios periféricos a través de un saco con líquido cefalorraquídeo (LCR).
El bebe requerirá de un tratamiento quirúrgico para cerrar el defecto, dentro de las primeras 36 horas de vida.
Los pacientes pueden evolucionar con grados variados de debilidad en miembros inferiores y disfunción vesical e intestinal, a pesar de la reparación quirúrgica temprana. En la mayoría se asocia a malformación de Chiari tipo II y a hidrocefalia, entre otras malformaciones.
La espina bífida oculta es un grupo de patologías heterogéneas con variada severidad, que presentan piel sana cubriendo el defecto.
Puede diagnosticarse tempranamente, por algún estigma en la piel en la zona de la columna (ej.: fosita, angiomas/manchas rojas, pelos, lipoma, bultos o apéndice). O tardíamente, al crecer el niño/a puede presentar clínica de médula anclada: debilidad progresiva en miembros, espasticidad, inestabilidad en la marcha, déficit sensorial, dolor en región dorsal o en miembros inferiores, escoliosis, atrofia muscular en miembros inferiores, deformidades en pies y/o alteraciones urológica (ej.: incontinencia urinaria/fecal, dificultad para el entrenamiento en inodoro, infecciones urinarias a repetición).
Dentro de este grupo se menciona:
Lipomielomeningocele, Lipoma medular, Filum terminale lipomatoso, Diastematomielia, diplomielia, Seno dérmico espinal, entre otros menos frecuentes.
Nuestro equipo esta especializado en estas complejas patologías, trabajando de forma integradora, en conjunto con otros especialistas de primer nivel, para mejorar la calidad del niño/a.
Atendemos en Centros de Salud con los mas modernos recursos tecnológicos para diagnóstico y tratamiento.
por NeCiP | May 28, 2025 | Malformación de Chiari Tipo I
El compromiso del paciente y su evolución dependerán del tipo de alteración.
Entre ellas se menciona:
Mielomeningocele, Espina bífida oculta, Encefaloceles, Diastematomielia,
Holoprosencefalia, Agenesia del cuerpo calloso, Lipomas cerebrales y medulares,
Displasia septo-óptica, Malformación de Dandy-Walker,
Microcefalia, hemimegalencefalia, Displasia cortical focal,
Lisencefalia, Heterotopias, Polimicrogiria, Esquizencefalia.
El asesoramiento genético es fundamental.
Nuestro equipo aborda al niño y niña de forma integradora, trabajando en conjunto con otros especialistas de primer nivel.
